[Text]
Genetiske analyser av aure i Gloppenelva
Rapportserie:
Rapport fra havforskningen 2023-19
ISSN: 1893-4536
Publisert: 24.03.2023
Forskningsgruppe(r):
Populasjonsgenetikk
Program:
Miljøeffekter av akvakultur
Approved by:
Research Director(s):
Geir Lasse Taranger
Program leader(s):
Terje Svåsand
English summary
1 - Introduksjon
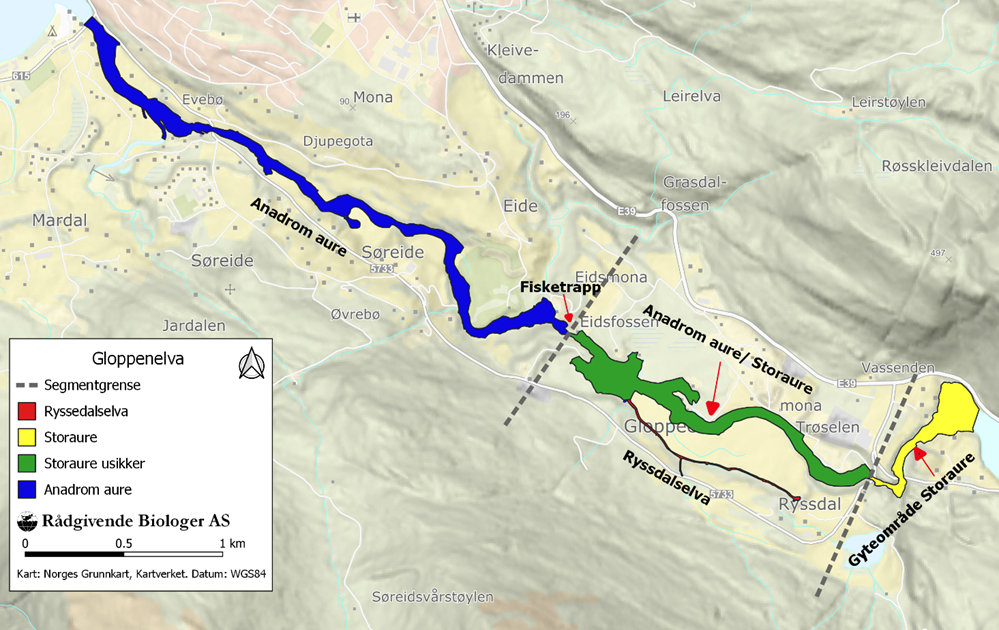
I Gloppenelva ble det åpnet en laksetrapp i Eidsfossen i 1972, men trappen ble stengt i 2014 etter store skader under en flomhendelse, og det er ikke avklart om trappen skal restaureres. Inntil 1972 var Eidsfossen vandringshinder for anadrom fisk i Gloppenvassdraget. Det er ikke kjent om sjøaure vandret opp trappen og reproduserte oppstrøms i perioden 1971 til 2014, og det er ikke gjennomført kultivering med sjøaure oppstrøms Eidsfossen eller flyttet sjøaure forbi fossen (Fig. 1). For å avklare om sjøaure har vandret opp og satt genetiske avtrykk i aurebestandene oppstrøms trappen i Eidsfossen ble det gjennomført genetiske analyser av aure på elvestrekningen mellom Eidsfossen og Trøselfossen som er neste vandringshinder oppstrøms, og i sideelva Ryssdalselva på denne strekningen. Aure fra dette området ble videre genetisk sammenlignet med sjøaure fanget under sportsfisket nedstrøms Eidsfossen og fra aure fanget på gyteområdet til storaure nær utløpet av Breimsvatnet oppstrøms Trøselfossen. Når det bygges en laksetrapp antas det i utgangspunktet at gytelaks- og sjøaure vandrer opp trappen og gyter i områdene ovenfor. Fisk som har vandret nedover som smolt er trolig mer motivert for å vandre opp trappen enn fisk som er født nedenfor. I tilfeller der trappeinngangen er vanskelig å finne på grunn av uheldig plassering kan manglende motivasjon for oppvandring medføre at kolonisering av områdene oppstrøms ikke skjer eller at det tar lang tid før produksjonspotensialet blir fullt utnyttet. Etter at laksetrappen ble åpnet i Eidsfossen i 1971 ble det årlig satt ut plommesekkyngel ovenfor trappen frem til 1995, men ved undersøkelser høsten 1995 ble det ikke registrert lakseunger på området. Dette tilsa at utsette lakseunger ikke eller i liten grad hadde overlevd. Det indikerte også at det ikke hadde vandret opp laks og gytt på strekningen eller at gytingen var mislykket. Med utgangspunkt i usikker oppvandring i laksetrappen og lite eller ikke tilslag bidrag fra utsettinger av plommesekkyngel ovenfor Eidsfossen ble det flyttet gytelaks oppom fossen høsten 1995 og det ble satt i gang utlegging av øyerogn av laks i Ryssdalselva vinteren 1997. Bakgrunnen var at laksesmolt som vandret ned fra området som smolt ville ha større motivasjon til å vandre opp trappen som voksne gytelaks og gyte ovenfor. Flyttingen av laks resulterte i vellykket gyting, men rekrutteringen var meget lav. Utleggingen av øyerogn vinteren 1997 og de etterfølgende årene medførte høy produksjon av laksesmolt i Ryssdalselva og mange vandret som åryngel ut i hovedelva ovenfor fossen og bidrog til en betydelig smoltproduksjon også her. I ettertid er det dokumentert oppvandring og vellykket gyting ved flere tilfeller etter 2000, trolig av fisk som var utsatt som egg på dette området og som passerte trappen under utvandring som smolt (Hellen & Urdal 2019).
2 - Materiale og metoder
2.1 - Prøvetaking
Skjell fra sjøaure fanget på anadrom strekning ble innsamlet under sportsfisket i fiskesesongen. Fra de øvrige områdene ble aureunger med alder 0 – 3 år fanget med elektrisk fiskeapparat på to faste stasjoner i hovedelva, på tre faste stasjoner i Ryssdalselva og på et større område på den øverste delen av gyteområdet i Breimsvatnet (Tabell 1, Fig. 1).
2.2 - DNA isolering og genotyping
DNA ble ekstrahert fra skjellprøver og finneklipp av innsamlet ungfisk med Qiagen DNeasyH96 Blood & Tissue Kit i 96-brønn plater, hver med to eller flere negative kontroller. Tjue-en genetiske markører (mikrosatelitter) ble analysert: Ssa410UOS, Ssa416UoS, Ssa408 (Cairney et al. 2000); SasaTAP2A, MHCI (Grimholt et al. 2002); Str2QUB, Str3QUB (Keenan et al. 2013); SsaD71 (King et al. 2005); mOne101, mOne102a&b (Olsen et al. 2000); Ssa197, Ssa85, (O'Reilly et al. 1996); Cocl-Lav-4 (Rogers et al. 2004); Oneμ9 (Scribner et al. 1996); SsOsl417 (Slettan et al. 1995); Ssa24NVH (Gharbi et al. 2006); Ssa87NVH (Moen et al. 2007); BS-131 (Presa & Guyomard 1996) og BG935488, CA048828, CA060177 (Vasemägi et al. 2005) i sammensatte multiplex (genotyping-betingelser tilgjengelig på forespørsel). PCR produkter ble analysert på en ABI 3730XL Genetic Analyser og fragmentstørrelsen i basepar (bp) ble beregnet med en 500LIZTM størrelsesstandard. Automatisk scorete alleler (varianter av mikrosatellittene) ble kontrollert manuelt av to forskere og, etter kvalitetskontroll, ble totalt 612 individer beholdt for videre statistiske analyser etter å ha akseptert maksimalt 12.5 % manglende genotyper. Programmet PGDSpider 2.1.1.3 (Lischer & Excoffier 2012) ble brukt til å utføre filkonvertering til forskjellige formater når det var nødvendig.
2.3 - Statistiske analyser
Totalt antall alleler og rikdom (Ar - også referert til som allelisk diversitet), dvs. en beregning av gjennomsnittlig antall alleler per locus når antall individer er korrigert for, ble beregnet med MSA (Dieringer & Schlötterer 2003). Antall private alleler (dvs. alleler som kun finnes i én prøve), observert (Ho) og forventet genetisk heterozygositet (uHe) ble beregnet med GenAlEx (Peakall & Smouse 2006). Genotypefordelingen av hvert locus per prøve og avvik fra forventningsverdi (heterozygot underskudd eller overskudd) ble sammenlignet med forventet antall avvik fra Hardy Weinberg ved hjelp av GENEPOP 7 (Rousset 2008) i tillegg til koblingsulikevekt (LD). Begge ble undersøkt ved bruk av følgende Markov-kjedeparametre: 10000 trinn av dememorisering, 1000 kjøringer og 10000 iterasjoner per kjøring. Signifikans ble vurdert gjennom Falsk Discovery Rate (FDR) (Benjamini & Hochberg 1995).
Populasjonsstrukturen ble undersøkt ved hjelp av ulike metoder. Først, ble hovedkomponentanalyse (PCA) utført ved bruk av funksjonen dudi.pca i ade4 (Dray & Dufour 2007). Manglende data ble i denne analysen erstattet med gjennomsnittlige allelfrekvenser, uten bruk av skalerte allelfrekvenser (skala = FALSE). Videre ble forholdet mellom prøvene undersøkt ved hjelp av diskriminant-analyse av hovedkomponenter (DAPC) (Jombart et al. 2010) implementert i programmet ADEGENET (Jombart 2008) hvor grupper ble definert ved hjelp av geografisk plassering av prøvene, innenfor ulike år. Kryssvalideringsfunksjon ble brukt for a beregne begge det optimale antallet hovedkomponenter og diskriminant-funksjoner som skal beholdes og derfor til å unngå overmontering av analysene (Jombart & Collins 2015; Miller et al. 2020). I tillegg, ble genetisk differensiering mellom prøver samlet inn på ulike lokaliteter testet ved bruk av parvis FST beregnet med programmet ARLEQUIN v.3.5.1.2. FST er et mål på populasjonsdifferensiering; dvs. genetisk avstand mellom populasjoner (forskjeller i allelfrekvens mellom to populasjoner) på grunn av genetisk struktur.
Større genetisk avstand mellom populasjoner betyr at det er liten utveksling av individer og liten genstrøm mellom dem. FST verdier varierer fra 0 til 1. Når verdien er null er det ingen differensiering, såkalt «panmixia», og alle individer kan i prinsippet krysse seg med alle andre individer. En FST verdi på 1 betyr at det ikke er noen utveksling av individer mellom populasjonene og de er fullstendig genetisk isolert fra hverandre. FST verdier mellom 0-0.05 har generelt blitt ansett for å indikere liten genetisk differensiering mens verdier mellom 0.05-0.25 indikerer moderat genetisk differensiering og verdier >0.25 betyr stor differensiering (Frankham et al. 2002). Men disse er omtrentlige retningslinjer fordi veldig lave FST verdier kan også representere viktige og biologisk betydningsfulle forskjeller (Knutsen et al. 2011).
Programmet STRUCTURE v. 2.3.4 ble brukt til å identifisere genetiske grupper i det innsamlede prøvematerialet. Dette programmet kan identifisere genetiske clustere/grupper i genetiske datasett ved å påvise allelfrekvensforskjeller i dataene. Programmet forsøker å finne den oppdelingen av individene i det antall genetiske clustere som gir høyest sannsynlighet. I analysen tester man et antall mulige clustere for å finne antallet grupperinger som er mest sannsynlig. STRUCTURE ble brukt med en modell som antar blanding og korrelerte allelfrekvenser uten å bruke populasjonsinformasjon i analysene. Analyser i STRUCTURE er beregningsintensive, og kjøringen ble derfor gjennomført med bruk av ParallelStructure (Besnier & Glover, 2013) noe som reduserer beregningstiden betydelig. Det ble gjort ti kjøringer med en innbrenningsperiode bestående av 100 000 replikasjoner og 1 000 000 MCMC-iterasjoner. Disse ble utført for K=1 til K=5 clustere. Resultatene fra disse analysene ble videre vurdert med to analytiske metoder for å beregne det optimale antall genetiske grupper som individene kunne deles inn i. For det første, ble ad hoc sammendragsstatistikk ΔK av Evanno et al. (2005) test beregnet. Denne testen er basert på endringshastigheten for «estimert sannsynlighet» mellom påfølgende K-verdier og tillater bestemmelse av det øverste hierarkiske strukturnivået i dataene. Analyser av resultatene fra STRUCTURE ble også videre analysert i STRUCTURESelector (Li & Liu 2018) som ble brukt for å beregne Puechmaille (2016)s fire statistikker (MedMed, MedMean, MaxMed og MaxMean) som er ansett som mer nøyaktig enn de tidligere brukte metodene for å bestemme det reelle antallet grupper. For de replikate kjøringene for den optimale verdien av k ble gjennomsnittet beregnet med CLUMPP v.1.1.1 (Jakobsson & Rosenberg 2007) ved hjelp av FullSearch algoritmen og illustrert grafisk som søylediagram. Resultater av analyser i STRUCTURE kan visualiseres som søylediagram hvor hver vertikal søyle representerer et individ, og fargene representerer tilhørighet til de ulike identifiserte clustere.
Analyser av slektskap mellom individene ble utført ved hjelp av COLONY v.2.0.5.1 (Jones & Wang 2010). I dette programmet benyttes sannsynlighetsmetoder for å vurdere mulig slektskap mellom individer, både mulige foreldre/avkom og eventuell hel- eller halvsøsken, basert på informasjon fra de genetiske markørene som er analysert. I analysen ble det tatt høyde for polygami (dvs. en fisk kan gyte med flere andre).
3 - Resultater og diskusjon
Totalt ble 612 individer samlet inn fra fire lokaliteter (Fig. 1) fra 1998 til 2019. Genetisk mangfold målt som allelisk rikdom eller heterozygositet var veldig lik på tvers av prøvene, men noe lavere i prøven fra Vassenden (Tabell 1). Derimot var antall private alleler størst i Vassenden, noe som indikerer liten sammenheng mellom Vassenden og de andre prøvelokalitetene.
| Lokalitet | År | Prøve | Antall individer | Antall alleler | Ar (Nmin=8) | Ar (Nmin=20) | Antall privat alleler | Ho | uHe | FIS | Avvik HWE (FDR) | Avvik LD (FDR) |
| Anadrom del, sjøaure | 1999 | GLO99S | 65 | 202 | 5.5 | 7.6 | 7 | 0.664 ± 0.036 | 0.703 ± 0.036 | 0.050 ± 0.018 | 2 (0) | 16 (2 |
| Anadrom del, sjøaure | 2018 | GLO18S | 15 | 147 | 5.6 | na | 2 | 0.689 ± 0.041 | 0.702 ± 0.042 | -0.023 ± 0.026 | 0 (0) | 2 (0) |
| Ryssdalselva | 1998 | RYS98 | 20 | 154 | 5.6 | 6.6 | 2 | 0.702 ± 0.044 | 0.711 ± 0.041 | -0.014 ± 0.024 | 1 (0) | 10 (0) |
| Ryssdalselva | 1999 | RYS99 | 22 | 168 | 5.8 | 7.8 | 2 | 0.707 ± 0.037 | 0.729 ± 0.036 | 0.002 ± 0.029 | 0 (0) | 29 (0) |
| Ryssdalselva | 2000 | RYS00 | 37 | 184 | 5.6 | 7.6 | 1 | 0.672 ± 0.042 | 0.699 ± 0.040 | 0.026 ± 0.020 | 3 (1) | 19 (0) |
| Ryssdalselva | 2009 | RYS09 | 16 | 139 | 5.3 | na | 0 | 0.649 ± 0.046 | 0.692 ± 0.037 | 0.039 ± 0.033 | 2 (1) | 19 (2) |
| Ryssdalselva | 2012 | RYS12 | 16 | 152 | 5.6 | na | 1 | 0.696 ± 0.044 | 0.698 ± 0.040 | -0.033 ± 0.037 | 3 (0) | 5 (0) |
| Ryssdalselva | 2014 | RYS14 | 28 | 167 | 5.4 | 7.3 | 0 | 0.689 ± 0.043 | 0.692 ± 0.041 | -0.024 ± 0.028 | 3 (1) | 19 (1) |
| Gloppenelva | 1998 | GLO98 | 44 | 183 | 5.5 | 7.4 | 3 | 0.690 ± 0.040 | 0.695 ± 0.042 | -0.014 ± 0.022 | 3 (1) | 7 (0) |
| Gloppenelva | 1999 | GLO99 | 91 | 203 | 5.4 | 7.4 | 3 | 0.671 ± 0.040 | 0.695 ± 0.040 | 0.031 ± 0.018 | 7 (2) | 18 (0) |
| Gloppenelva | 2000 | GLO00 | 24 | 159 | 5.3 | 7.2 | 0 | 0.686 ± 0.047 | 0.680 ± 0.042 | -0.024 ± 0.027 | 2 (1) | 10 (0) |
| Gloppenelva | 2009 | GLO09 | 29 | 175 | 5.6 | 7.6 | 0 | 0.664 ± 0.046 | 0.692 ± 0.044 | 0.025 ± 0.028 | 5 (1) | 7 (1) |
| Gloppenelva | 2011 | GLO11 | 60 | 193 | 5.5 | 7.4 | 3 | 0.685 ± 0.045 | 0.701 ± 0.041 | 0.017 ± 0.021 | 3 (1) | 24 (1) |
| Gloppenelva | 2012 | GLO12 | 15 | 153 | 5.7 | na | 3 | 0.690 ± 0.052 | 0.702 ± 0.046 | -0.016 ± 0.031 | 1 (0) | 5 (0) |
| Gloppenelva | 2014 | GLO14 | 8 | 110 | 5.2 | na | 0 | 0.726 ± 0.041 | 0.714 ± 0.033 | -0.091 ± 0.043 | 0 (0) | 1 (0) |
| Gloppenelva | 2019 | GLO19 | 67 | 199 | 5.5 | 7.5 | 3 | 0.697 ± 0.040 | 0.703 ± 0.041 | -0.004 ± 0.015 | 5 (4) | 17 (2) |
| Vassenden | 2017 | VASS17 | 55 | 165 | 4.8 | 6.4 | 13 | 0.636 ± 0.047 | 0.640 ± 0.047 | -0.005 ± 0.018 | 4 (2) | 13 (0) |
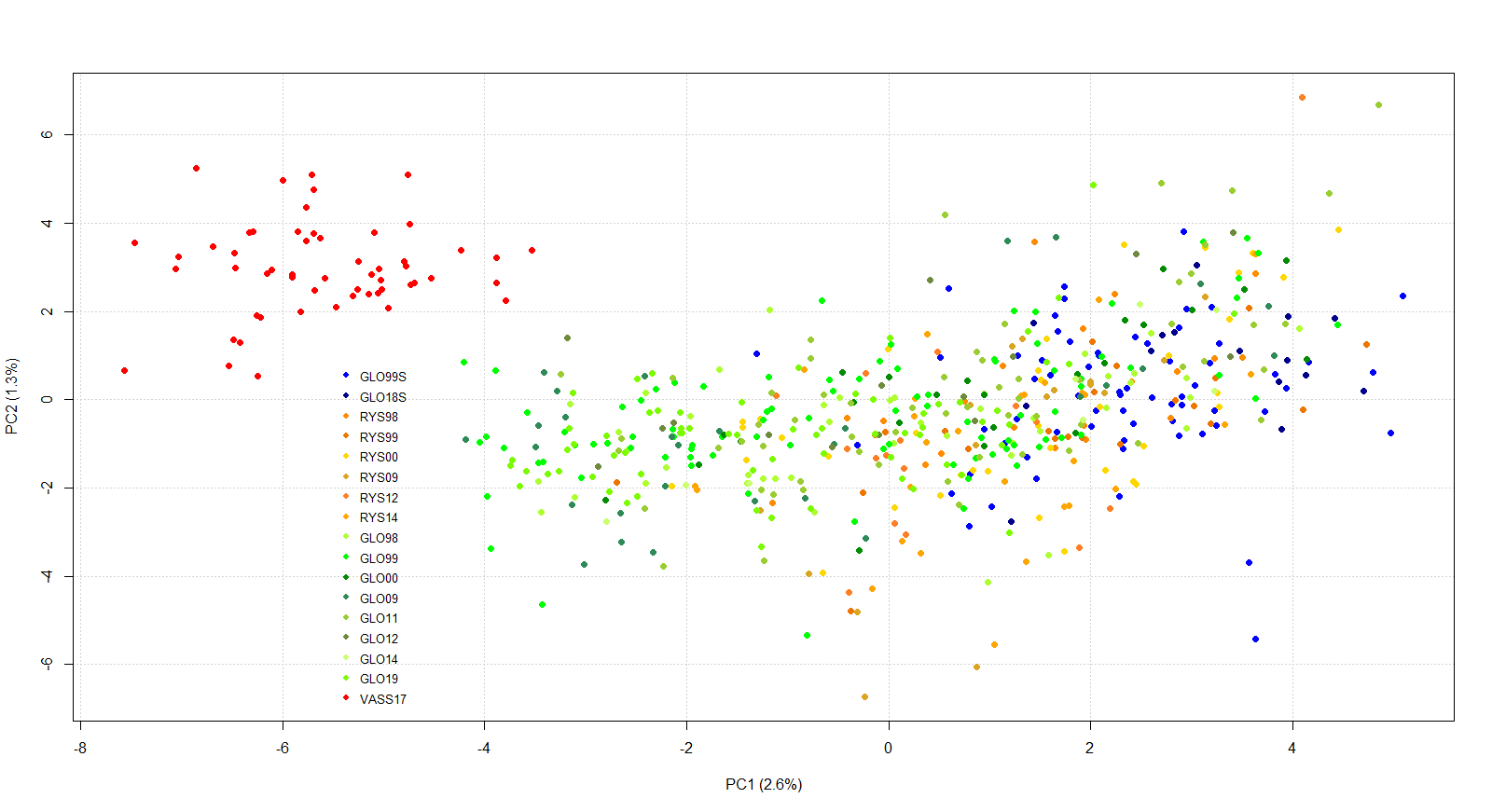
Den første aksen av hovedkomponentanalysen (PCA) forklarte 2.6% av variasjonen og indikerte at Vassenden er isolert fra de andre prøvene som overlappet til en viss grad (Fig. 2). Den andre aksen (1.3%) bidro ikke så mye bortsett fra å støtte observasjonen fra den første aksen.
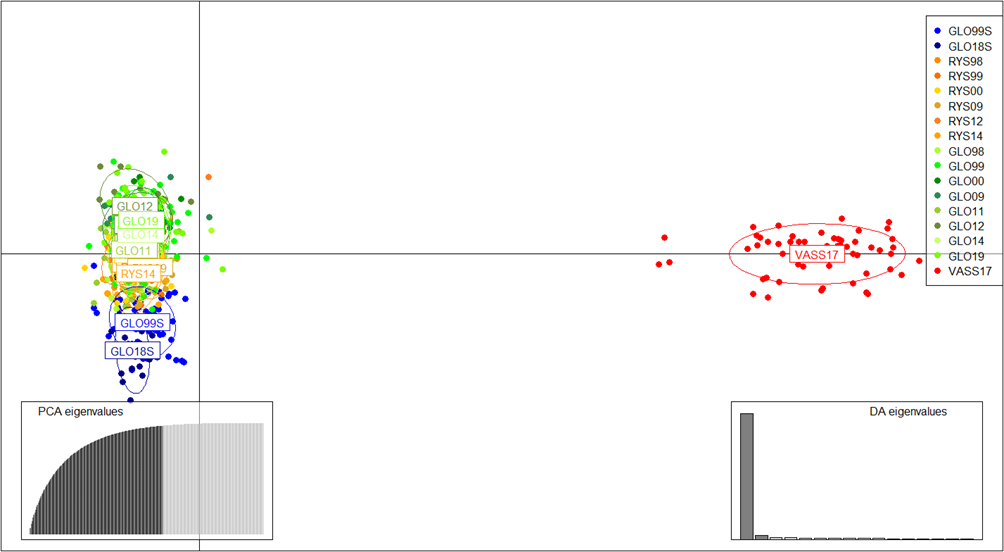
I diskrimininantanalysen av hovedkomponenter (DAPC) utført med alle individer (Fig. 3a) viste at den første aksen, som forklart hele 88.5% av variasjonen, at Vassenden skilte seg klart ut fra de andre lokalitetene, noe som antyder genetisk isolasjon og liten utveksling av individer mellom denne lokaliteten og de andre lokalitetene nedstrøms. I de påfølgende analyser ble derfor Vassenden utelatt for å bedre illustrere forholdet mellom sjøørret, Ryssdalselva og Gloppenelva.
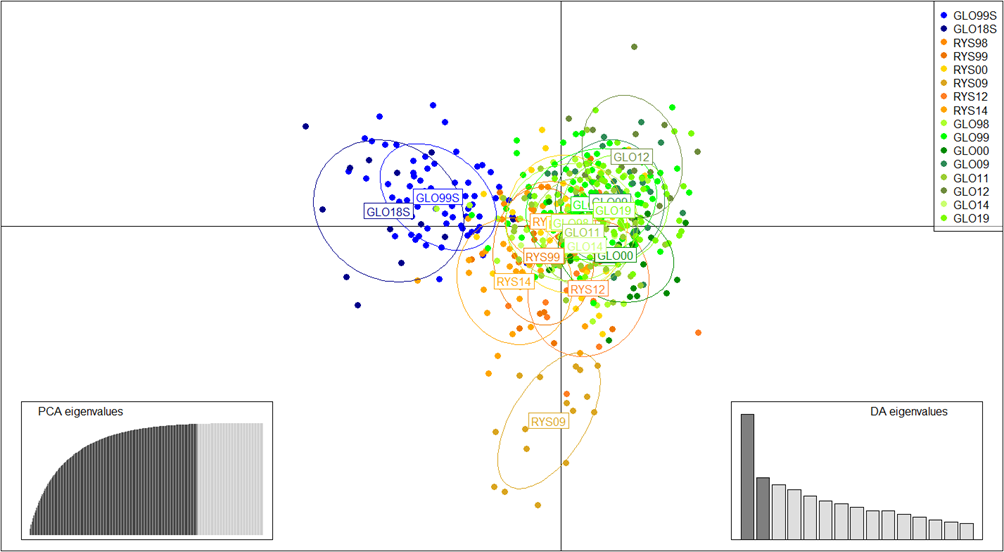
I det «trimmede» datasettet (Fig. 3b) separerte den første aksen av differensiering (20.8% av variasjonen) sjøørret fra de to andre lokalitetene som overlappet til en viss grad. Den andre aksen (10.3% av variasjonen) isolerte individer samlet i Ryssdalselva i 2009 fra resten. Med bakgrunn i mengden variasjon som hovedaksene i denne grafen forklarer, kan vi konkludere at det er en viss sammenheng mellom sjøørreten, Gloppenelva og Ryssdalselva, men det er også genetiske forskjeller mellom disse lokalitetene.
Parvise FST analyser ble utført mellom de 17 prøvene men noen av dem hadde få individer (N<25) og derfor lavere statistisk styrke (fremhevet i grå farge i Tabell 2). Den største differensiering ble observert mellom Vassenden og de andre prøvene, spesielt når Vassenden ble sammenlignet med sjøørret (særlig prøven fra 2018) mens den laveste differensieringen ble observert mellom Ryssdalselva og Gloppenelva. Prøvene fra Ryssdalselva viste ingen intern differensiering unntatt mellom de eldste prøvene (samlet inn i 1998-1999) versus de nyeste (2014). I Gloppenelva, var det forskjeller mellom den nyeste prøven (2019) og prøvene som var samlet inn i 1999, 2000, 2011 og 2014, men det var bare prøvene fra 2000 og 2009 som var signifikant forskjellig. Prøvene av sjøørret, samlet over to tiår, viste ikke noen differensiering. Når man sammenligner sjøørret med resten av prøvene, viste den parvise FST matrisen at sjøørret var signifikant forskjellig fra alle prøvene i Gloppenelva med unntak av GLO14, og også signifikant forskjellig fra alle prøvene fra Ryssdalselva som ble samlet etter 1999. Sammenligninger mellom alle de ulike prøvene fra Ryssdalselva og Gloppenelva viste alle lav og ikke signifikant differensiering.
| GLO99S | GLO18S | RYS98 | RYS99 | RYS00 | RYS09 | RYS12 | RYS14 | GLO98 | GLO99 | GLO00 | GLO09 | GLO11 | GLO12 | GLO14 | GLO19 | VASS17 | |
| GLO99S | * | 0.115 | 0.020 | 0.038 | 0.000 | 0.000 | 0.006 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.021 | 0.000 | 0.000 |
| GLO18S | 0.005 | * | 0.016 | 0.020 | 0.009 | 0.001 | 0.000 | 0.003 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.008 | 0.000 | 0.000 |
| RYS98 | 0.007 | 0.013 | * | 0.643 | 0.272 | 0.017 | 0.663 | 0.001 | 0.008 | 0.010 | 0.060 | 0.003 | 0.027 | 0.501 | 0.022 | 0.000 | 0.000 |
| RYS99 | 0.006 | 0.012 | 0.000 | * | 0.323 | 0.248 | 0.334 | 0.007 | 0.003 | 0.000 | 0.007 | 0.000 | 0.001 | 0.171 | 0.259 | 0.000 | 0.000 |
| RYS00 | 0.008 | 0.012 | 0.002 | 0.002 | * | 0.020 | 0.446 | 0.011 | 0.045 | 0.001 | 0.352 | 0.005 | 0.010 | 0.193 | 0.260 | 0.000 | 0.000 |
| RYS09 | 0.018 | 0.026 | 0.015 | 0.005 | 0.012 | * | 0.251 | 0.097 | 0.005 | 0.000 | 0.006 | 0.005 | 0.000 | 0.005 | 0.510 | 0.000 | 0.000 |
| RYS12 | 0.010 | 0.023 | 0.000 | 0.002 | 0.001 | 0.005 | * | 0.246 | 0.755 | 0.543 | 0.349 | 0.541 | 0.393 | 0.603 | 0.285 | 0.169 | 0.000 |
| RYS14 | 0.013 | 0.015 | 0.016 | 0.012 | 0.008 | 0.008 | 0.003 | * | 0.001 | 0.000 | 0.004 | 0.000 | 0.000 | 0.001 | 0.056 | 0.000 | 0.000 |
| GLO98 | 0.016 | 0.024 | 0.009 | 0.011 | 0.004 | 0.012 | 0.000 | 0.012 | * | 0.825 | 0.060 | 0.599 | 0.300 | 0.310 | 0.230 | 0.060 | 0.000 |
| GLO99 | 0.023 | 0.032 | 0.008 | 0.013 | 0.007 | 0.018 | 0.000 | 0.017 | 0.000 | * | 0.022 | 0.486 | 0.013 | 0.900 | 0.219 | 0.000 | 0.000 |
| GLO00 | 0.021 | 0.026 | 0.006 | 0.011 | 0.001 | 0.015 | 0.001 | 0.011 | 0.004 | 0.006 | * | 0.006 | 0.032 | 0.476 | 0.076 | 0.000 | 0.000 |
| GLO09 | 0.030 | 0.037 | 0.014 | 0.021 | 0.010 | 0.017 | 0.000 | 0.018 | 0.000 | 0.000 | 0.011 | * | 0.088 | 0.370 | 0.104 | 0.353 | 0.000 |
| GLO11 | 0.017 | 0.022 | 0.007 | 0.012 | 0.006 | 0.016 | 0.001 | 0.011 | 0.001 | 0.004 | 0.005 | 0.004 | * | 0.441 | 0.545 | 0.000 | 0.000 |
| GLO12 | 0.021 | 0.026 | 0.000 | 0.006 | 0.004 | 0.022 | 0.000 | 0.017 | 0.001 | 0.000 | 0.000 | 0.002 | 0.001 | * | 0.118 | 0.800 | 0.000 |
| GLO14 | 0.015 | 0.023 | 0.018 | 0.006 | 0.005 | 0.001 | 0.004 | 0.013 | 0.003 | 0.005 | 0.010 | 0.011 | 0.000 | 0.010 | * | 0.011 | 0.000 |
| GLO19 | 0.033 | 0.043 | 0.013 | 0.020 | 0.016 | 0.023 | 0.003 | 0.020 | 0.003 | 0.005 | 0.015 | 0.001 | 0.007 | 0.000 | 0.017 | * | 0.000 |
| VASS17 | 0.105 | 0.121 | 0.090 | 0.102 | 0.089 | 0.091 | 0.075 | 0.091 | 0.064 | 0.062 | 0.083 | 0.054 | 0.068 | 0.064 | 0.076 | 0.055 | * |
Testene utført etter STRUCTURE analysen konkluderte med at K=4 var det det mest sannsynlige antallet genetiske grupper (clusters). Strukturdiagramet (Fig. 4) bekreftet fraværet av kobling mellom Vassenden og alle de andre prøvene. Analysen viser også at enkelte individer med genetisk profil som ligner på sjøaure påvises i Gloppenelva og Ryssdalselva, særlig i prøvene fra perioden fram til 2014. Et interessant resultat er den lave grad av innblanding funnet på individnivå: 441 individer (72%) viste en grad av tilhørighet til genetisk gruppe ≥0.8; dvs. andelen individer som tilhører bare én genetisk gruppe er over 80%.
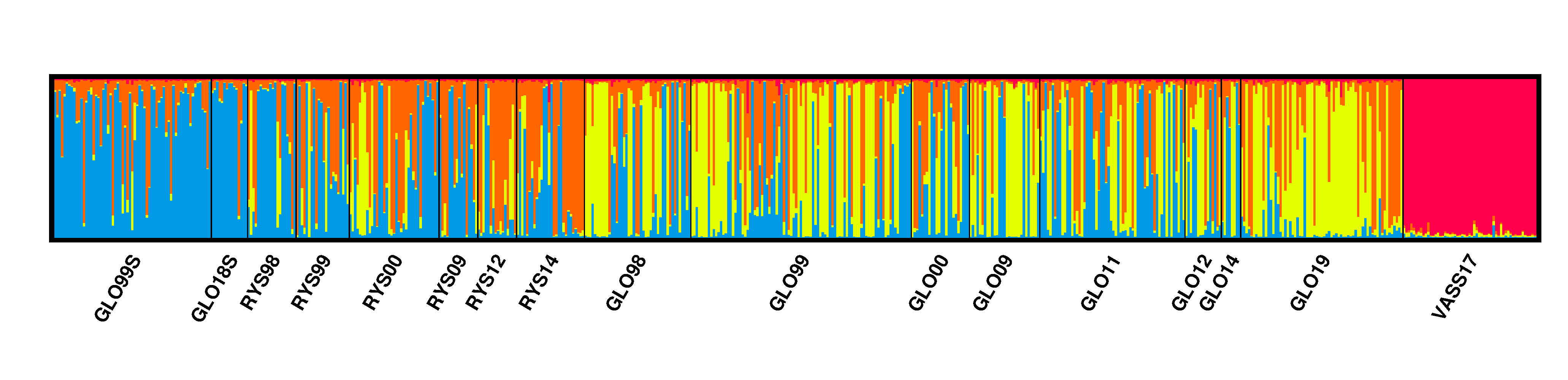
COLONY identifiserte 38 familier med helsøsken, 26 av dem med sannsynlighet høyere enn 0.8. Fire av disse familiene bestod av individer fra forskjellige lokaliteter og helsøsken ble funnet bare mellom Gloppenelva og Ryssdalselva. Ingen helsøskenpar ble identifisert i prøvene av sjøørret, og bare ett i Vassenden (Tabell 3). Individer klassifisert som tilhørende samme familie ble samlet inn enten i samme år eller påfølgende år. Et helsøskenpar ble funnet i Ryssdalselva mellom individer samlet inn i 2009 og 2012; og en annen familie, også fra Ryssdalselva, bestod av individer samlet i 2009, 2012 og 2014. Mangelen på helsøsken mellom Vassenden og de resterende prøvene bekrefter at det er liten utveksling av individer mellom disse lokalitetene.
| Medlem1 | Medlem2 | Medlem3 | Medlem4 | Medlem5 | Sanns (Ink.) |
| GLO99_377 | GLO00_247 | 1.0000 | |||
| RYS00_323 | RYS00_325 | RYS00_407 | GLO99_417 | GLO00_256 | 1.0000 |
| RYS99_95 | RYS99_96 | 1.0000 | |||
| GLO11_123 | GLO11_207 | 0.9998 | |||
| RYS09_402 | RYS09_445 | 0.9998 | |||
| RYS14_190 | RYS14_212 | RYS14_214 | 0.9998 | ||
| GLO11_106 | GLO11_15 | GLO11_18 | 0.9997 | ||
| RYS14_166 | RYS14_213 | 0.9997 | |||
| GLO99_345 | GLO99_371 | GLO99_393 | 0.9996 | ||
| RYS99_12 | RYS09_442 | RYS14_209 | 0.9996 | ||
| RYS98_30 | RYS99_31 | 0.9987 | |||
| RYS98_33 | RYS99_72 | 0.9977 | |||
| RYS09_342 | RYS09_412 | 0.9930 | |||
| RYS12_61 | GLO11_202 | 0.9921 | |||
| GLO99_361 | GLO99_416 | GLO00_254 | 0.9902 | ||
| GLO19_40 | GLO19_51 | 0.9898 | |||
| RYS09_328 | RYS12_39 | 0.9869 | |||
| RYS99_27 | GLO99_307 | 0.9853 | |||
| GLO99_303 | GLO99_309 | 0.9799 | |||
| GLO11_23 | GLO11_73 | 0.9779 | |||
| RYS98_7 | RYS98_75 | GLO98_144 | GLO99_378 | 0.9649 | |
| RYS09_443 | RYS09_446 | 0.9644 | |||
| GLO19_64 | GLO19_95 | 0.8972 | |||
| RYS99_13 | RYS99_42 | 0.8648 | |||
| GLO99_392 | GLO99_410 | 0.8365 | |||
| VASSENDEN079 | VASSENDEN119 | 0.8155 |
4 - Konklusjoner
Vassenden er genetisk isolert fra alle de andre prøvene som ble analysert. Vi konkluderer derfor med at fisk i denne delen av vassdraget ikke er knyttet til elvedelene nedstrøms i særlig grad. Vi kan ikke utelukke at en og annen fisk kan ha sluppet seg nedover og dermed bidratt til bestandene nedstrøms. Dette skjer sannsynligvis sjeldent, gitt de relativt store genetiske forskjellene som er observert mellom individer fra Vassenden og resten av vassdraget.
Selv om Gloppenelva, Ryssdalselva og sjøørretprøvene ser ut til å ha en viss grad av genetisk overlapp, kan disse delene av vassdraget likevel ikke betraktes som en homogen genetisk enhet (eller en homogen populasjon). Det finnes genetiske forskjeller mellom prøvene av sjøørret og prøvene samlet inn fra Gloppenelva og Ryssdalselva som Figur 3b viser. Likevel viser clusteranalysen (Fig. 4) at enkelte sjøørret kan ha brukt området ovenfor fisketrappa som gyteområde og dermed påvirket bestandene her genetisk.
5 - Referanser
Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289-300.
Cairney M, Taggart JB, HØyheim B (2000) Characterization of microsatellite and minisatellite loci in Atlantic salmon ( Salmo salar L.) and cross-species amplification in other salmonids. Molecular Ecology 9, 2175-2178.
Dieringer D, Schlötterer C (2003) MICROSATELLITE ANALYSER (MSA): A platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes 3, 167-169.
Dray S, Dufour A-B (2007) The ade4 Package: Implementing the Duality Diagram for Ecologists. Journal of Statistical Software 22, 1 - 20.
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to Conservation Genetics Cambridge University Press, Cambidge.
Gharbi K, Gautier A, Danzmann RG, et al. (2006) A linkage map for brown trout ( Salmo trutta ): chromosome homeologies and comparative genome organization with other salmonid fish. Genetics 172, 2405-2419.
Grimholt U, Drabløs F, Jørgensen S, Høyheim B, Stet R (2002) The major histocompatibility class I locus in Atlantic salmon ( Salmo salar L.): polymorphism, linkage analysis and protein modelling. Immunogenetics 54, 570-581.
Hellen BA, Urdal K (2019) Fiskeundersøkelser i Gloppenelva i 2018. Rådgivende Biologer AS, rapport 2885, 20 sider, ISBN 978-82-8308-619-5.
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801-1806.
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403-1405.
Jombart T, Collins C (2015) A tutorial for discriminant analysis of principal components (DAPC) using adegenet 2.0.0.
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 11, 94.
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Molecular Ecology Resources 10, 551-555.
Keenan K, Bradley CR, Magee JJ, et al. (2013) Beaufort trout MicroPlex: a high-throughput multiplex platform comprising 38 informative microsatellite loci for use in resident and anadromous (sea trout) brown trout Salmo trutta genetic studies. Journal of Fish Biology 82, 1789-1804.
King TL, Eackles MS, Letcher BH (2005) Microsatellite DNA markers for the study of Atlantic salmon (Salmo salar) kinship, population structure, and mixed-fishery analyses. Molecular Ecology Notes 5, 130-132.
Knutsen H, Olsen EM, Jorde PE, et al. (2011) Are low but statistically significant levels of genetic differentiation in marine fishes ‘biologically meaningful’? A case study of coastal Atlantic cod. Molecular Ecology 20, 768-783.
Li Y-L, Liu J-X (2018) StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Molecular Ecology Resources 18, 176-177.
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298-299.
Miller JM, Cullingham CI, Peery RM (2020) The influence of a priori grouping on inference of genetic clusters: simulation study and literature review of the DAPC method. Heredity 125, 269-280.
Moen T, Sonesson AK, Hayes B, et al. (2007) Mapping of a quantitative trait locus for resistance against infectious salmon anaemia in Atlantic salmon ( Salmo salar ): comparing survival analysis with analysis on affected/resistant data. BMC Genet 8, 53.
O'Reilly PT, Hamilton LC, McConnell SK, Wright JM (1996) Rapid analysis of genetic variation in Atlantic salmon ( Salmo salar ) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Canadian Journal of Fisheries and Aquatic Sciences 53, 2292-2298.
Olsen JB, Wilson SL, Kretschmer EJ, Jones KC, Seeb JE (2000) Characterization of 14 tetranucleotide microsatellite loci derived from sockeye salmon. Molecular Ecology 9, 2185-2187.
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6, 288-295.
Presa P, Guyomard R (1996) Conservation of microsatellites in three species of salmonids. Journal of Fish Biology 49, 1326-1329.
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Molecular Ecology Resources 16, 608-627.
Rogers SM, Marchand M-H, Bernatchez L (2004) Isolation, characterization and cross-salmonid amplification of 31 microsatellite loci in the lake whitefish ( Coregonus clupeaformis , Mitchill). Molecular Ecology Notes 4, 89-92.
Rousset F (2008) GENEPOP'007: a complete re-implementation of the genepop software for Windows and Linux. Molecular Ecology Resources 8, 103-106.
Scribner KT, Gust JR, Fields RL (1996) Isolation and characterization of novel salmon microsatellite loci: Cross-species amplification and population genetic applications. Canadian Journal of Fisheries and Aquatic Sciences 53, 833-841.
Slettan A, Olsaker I, Lie Ø (1995) Atlantic salmon, Salmo salar , microsatellites at the SSOSL25, SSOSL85, SSOSL311, SSOSL417 loci. Animal Genetics 26, 281-282.
Vasemägi A, Nilsson J, Primmer CR (2005) Expressed Sequence Tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon ( Salmo salar L.). Molecular Biology and Evolution 22, 1067-1076.